New molecular mechanisms revealed towards cracking the combinatorial “histone code”
Recently, Dr. Haitao Li group in Center for Structural Biology and Tsinghua Medical School published two research papers online in Nature (March 02, 2014) and Genes & Dev (March 03, 2014), reporting on the structural basis for histone methylation pattern readout by ZMYND11 and Spindlin1, respectively. The two studies revealed new mechanistic insights into “histone methylation” decoding, and further unveil the mystery of epigenetic regulation.
In eukaryotes, gene expression is not simply controlled by specific DNA elements; it is also under exquisite regulation by histone post-translational modifications (PTMs). To date, more than 25 distinct types of histone PTMs have been identified, such as methylation, acylation and phosphorylation and so on. These histone PTMs or their combination were loosely defined as a “histone code” that plays a major role in epigenetic regulation. Besides, histones (H1, H2A, H2B, H3 and H4) often exist in multiple forms of sequence variants. Histone PTMs and histone variants significantly expand the indexing potential of histones, and contribute to the formation of an additional layer of epigenetic information beyond DNA. Accumulating evidence showed that dysregulation of “histone code” is often linked to human disease, notably cancer. Thus, studying the regulatory mechanisms underlying “writing”, “reading” and “erasing” of particular histone PTM are of significant value to the understanding of human disease and its therapy.
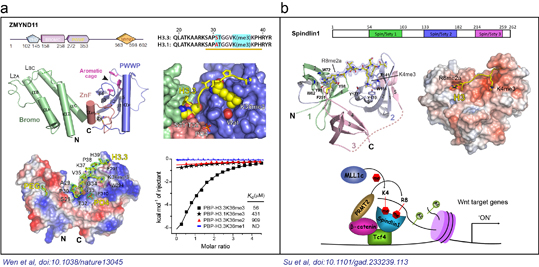
Fig. 1, a) Histone variant H3.3 lysine 36 trimethylation readout by tandem Bromo-ZnF-PWWP domains of ZMYND11; b) Histone H3 lysine-arginine methylation pattern readout by Spin/Ssty repeats of Spindlin1.
The paper published in Nature illustrated the molecular basis on how a candidate tumor suppressor, ZMYND11, recognizes histone variant H3.3K36me3 modification with its tandem Bromo-ZnF-PWWP domains (Fig. 1a). Functional and bioinformatic analysis further confirmed a role of ZMYND11 in tumor suppression by refraining oncogene expression at transcription elongation level. This paper reported for the first time a histone variant-specific lysine methylation “reader”. Such dual recognition of histone variant as well as methylation type represents a true highlight in recent histone researches, thus underscoring the importance and complexity of eukaryotic epigenetic regulation. This paper is a joint effort from Dr. Haitao Li laboratory at Tsinghua University, Dr. Xiaobing Shi laboratory at University of Texas MD Anderson Cancer Center, and Dr. Wei Li laboratory at Baylor College of Medicine. The whole paper covers in-depth researches in the fields of structural biology, cellular biology and functional genomics, highlighting the importance of multi-disciplinary collaboration in current life science research. Dr. Haitao Li and his postdoctoral fellow, Yuanyuan Li, hold the co-correspondence and co-first authorship of this paper, respectively.
The paper published in Genes & Dev reported the molecular basis underlying histone H3 “K4me3-R8me2a” methylation pattern readout by human Spindlin1 (Fig. 1b). Biochemistry and cell-based functional assays established a critical role of this histone methylation pattern readout in Wnt target gene expression at the transcriptional level. Spindlin1, a member of Spin/Ssty family proteins, is often highly expressed in various cancers and participates in the regulation of cell cycle and transcription. Structural studies showed that Spindlin1 specifically recognizes H3 “K4me3-R8me2a” pattern through its second and first Spin/Ssty repeats. Calorimetric titration measured a binding constant of ~45 nM, representing the strongest binding event among reported histone “mark-reader” pairs. Dr. Haiao Li from School of Medicine and Dr. Wei Wu from School of Life Sciences are corresponding authors of this paper; Xiaonan Su, a 2010 PhD student from School of Medicine and Guixin Zhu, a postdoctoral fellow from School of Life Sciences shared the co-first authorship.
Dr. Haitao Li joined Tsinghua School of Medicine as a full-time faculty in January 2010. He has been working in the field of structural epigenetics for nearly ten years. In 2007, Dr. Li and his collaborator, Prof. C. David Allis at The Rockefeller University, proposed the hypothesis of multivalent histone PTMs readout by paired “reader” modules (Rev Mol Cell Biol 8: 983-994). To testify this hypothesis, Dr. Li group published one paper on human ATRX recognition of H3“K4me0-K9me3” pattern (Nat Struct Mol Biol 18: 769-776) and another on human BPTF recognition of “H3K4me3-H4K16ac” at nucleosome level (Cell 145: 692-706) in 2011. The two recent works reported here represent new breakthroughs that Dr. Li group has made in the past two years, towards cracking the combinatorial “histone code”. It is believed that these studies not only help to elucidate the relationship between “histone code” and human disease, but also pave the way for structure-based epigenetic drug discovery in the future.
The above researches were supported by grants from the MOST “973” project, MOE NCET talent program, NSFC foundation, Tsinghua “221” talent program and Tsinghua-Peking Center for Life Sciences. Shanghai Synchrotron Radiation Facility (SSRF) and Beijing Synchrotron Radiation Facility (BSRF) provided supports for data collection.
Link:
1)http://www.nature.com/nature/journal/vaop/ncurrent/full/nature13045.html
2)http://genesdev.cshlp.org/cgi/doi/10.1101/gad.233239.113